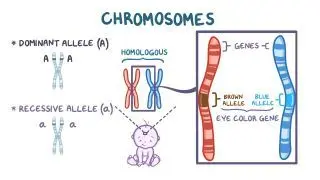
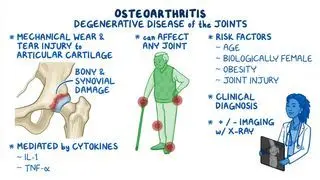
Harel T, Rabinowitz R, Hendler N, et al. COL11A2 mutation associated with autosomal recessive Weissenbacher-Zweymuller syndrome: molecular and clinical overlap with otospondylomegaepiphyseal dysplasia (OSMED). Am J Med Genet A. 2004;132(1):33-35. https://doi.org/10.1002/ajmg.a.30371
Otospondylomegaepiphyseal dysplasia. Genetic and Rare Diseases Information Center. Published April 13, 2023. Accessed April 13, 2023. https://rarediseases.info.nih.gov/diseases/4130/osmed-syndrome
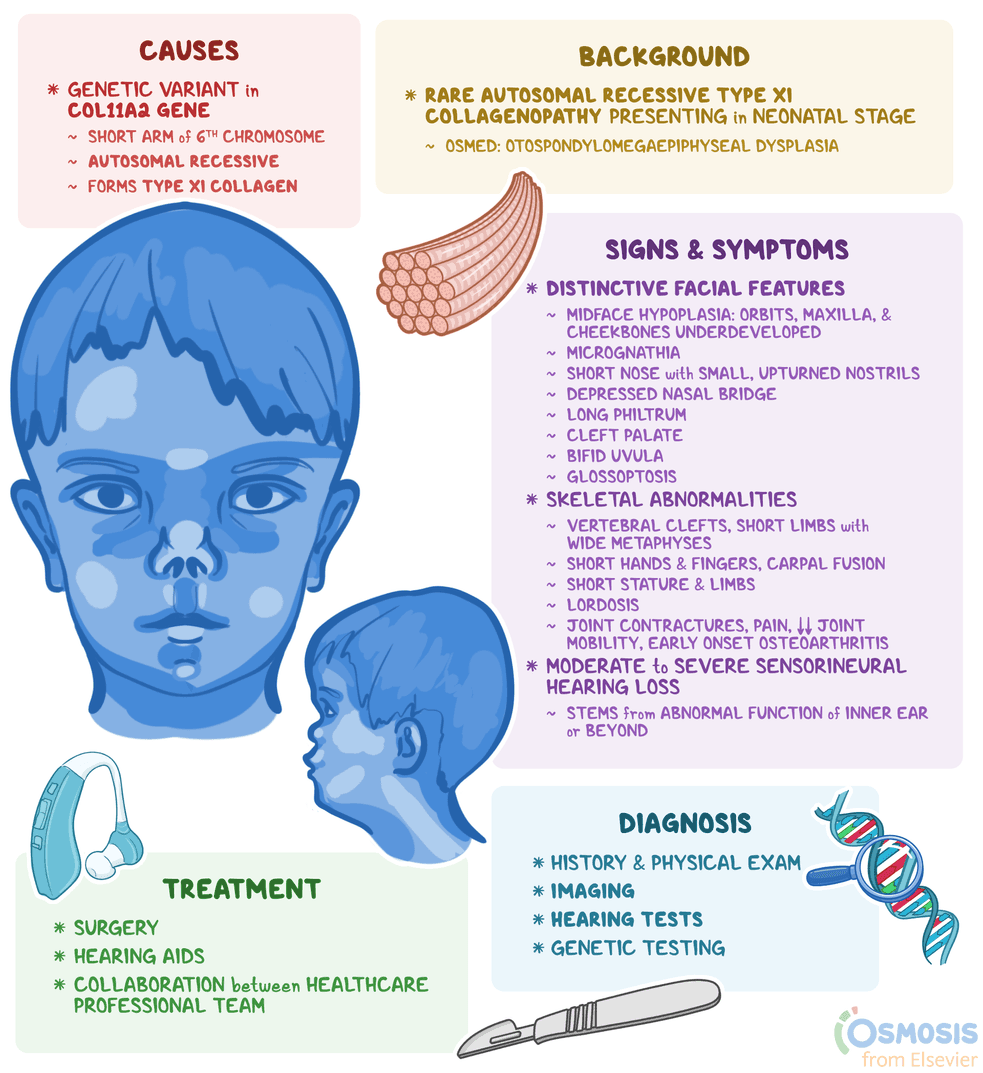
Otospondylomegaepiphyseal dysplasia. Orphanet. Published April 13, 2023. Accessed April 13, 2023. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=1427
Otospondylomegaepiphyseal dysplasia, autosomal dominant; osmeda. Online Mendelian Inheritance in Man (OMIM). Accessed November 22, 2025. https://www.omim.org/entry/184840?search=col11a2&highlight=col11a2
Stickler syndrome, type II; STL2. Online Mendelian Inheritance in Man (OMIM). Accessed November 22, 2025. https://www.omim.org/entry/604841
Temtamy SA, Männikkö M, Abdel-Salam GMH, et al. Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical and radiological findings in sibs homozygous for premature stop codon mutation in the COL11A2 gene. Am J Med Genet A. 2006;140(11):1189-1195. https://doi.org/10.1002/ajmg.a.31205